How Useful Are Common Simulants of Chemical Warfare Agents at Predicting Adsorption Behavior?
Abstract
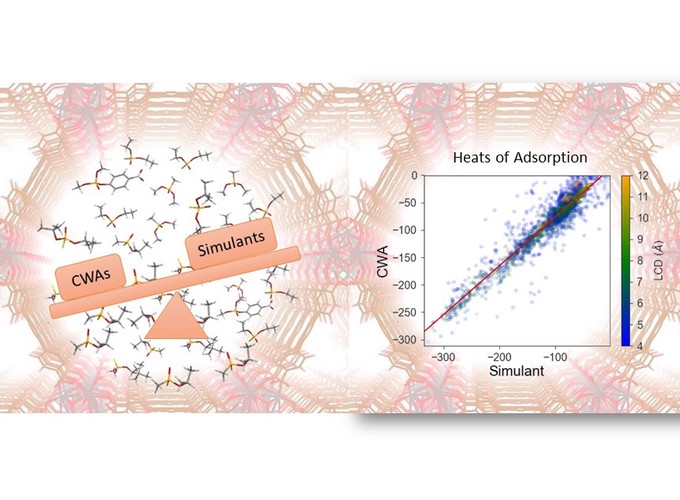
Nanoporous materials such as metal–organic frameworks (MOFs) have attractive properties for selective capture of chemical warfare agents (CWAs). For obvious reasons, most research on adsorption of CWAs is performed with simulant molecules rather than real agents. This paper examines how effectively common CWA simulants mimic the adsorption properties of sarin and soman. To this end, we perform molecular simulations in the dilute adsorption limit for four simulants [dimethyl methylphosphonate (DMMP), diethyl chlorophosphate (DCP), diisopropyl fluorophosphate, and dimethyl p-nitrophenyl phosphate (DMNP)] and sarin and soman in a set of 2969 MOFs with experimentally known crystal structures. To establish the robustness of the conclusions with respect to the force field used in these simulations, each system was examined with two independent force fields, a “generic” force field and a density functional theory (DFT)-derived force field we established based on extensive dispersion-corrected DFT calculations of adsorption in the well-known MOF UiO-66. Our results show that when judging the performance of adsorbents using the heat of adsorption, DCP and DMMP are the best simulants for the adsorption of sarin, while DMNP is the best simulant for soman. The adsorption properties of DCP or DMMP show a strong correlation with sarin over a range of MOFs, but the correlation between DMNP and soman is considerably weaker. Comparisons of results with both force fields indicate that our main conclusions are robust with respect to the force field used to define adsorbate–MOF interactions.
Mayank Agrawal
Postdoctoral Research Associate
My research include atomistic simulation of nano-porous materials for separation and catalysis.