Effect of Intrinsic Flexibility on Adsorption Properties of Metal-Organic Frameworks at Dilute and Non-dilute Loadings
Abstract
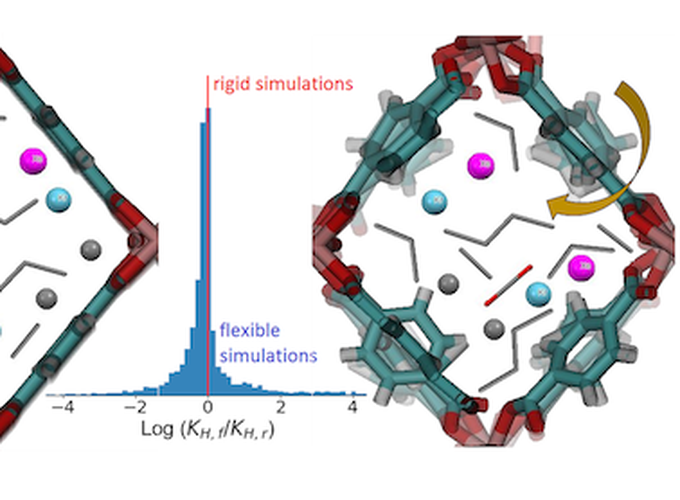
Molecular simulation of adsorption in nanoporous materials has become a valuable complement to experimental studies of these materials. In almost all cases, these simulations treat that adsorbing material as rigid. We use molecular simulations to examine the validity of this approximation for the adsorption in metal-organic frameworks (MOFs) that have framework flexibility without change in their unit cells due to thermal vibrations. All nanoporous materials are subject to this kind of framework flexibility. We examine the adsorption of 9 molecules (CO2, CH4, ethane, ethene, propane, propene, butane, Xe and Kr) and four molecular mixtures (CO2/CH4, ethane/ethene, propane/propene/butane, and Xe/Kr) in 100 MOFs at dilute and non-dilute adsorption conditions. Our results show that single component adsorption uptakes at non-dilute conditions are only weakly affected by framework flexibility, but adsorption selectivities at both dilute and non-dilute conditions can be significantly affected by flexibility. The most dramatic impacts of framework flexibility occur for adsorption uptake in the limit of dilute adsorption. These results suggest that the importance of including framework flexibility when attempting to make quantitative predictions of adsorption selectivity in MOFs and similar materials may have been underestimated in the past.
Mayank Agrawal
Postdoctoral Research Associate
My research include atomistic simulation of nano-porous materials for separation and catalysis.